Location:Home >> Detail
Med One. 2016; 1(4): 6; https://doi.org/10.20900/mo.20160019

1 Department of Hematology, Xinqiao Hospital, Third Military Medical University, Xinqiao, Chongqing 400037, P.R. China
2 National Institute of Biomedical Imaging and Bioengineering (NIBIB), NIH, Bethesda, MD 20892, USA
3 National Heart, Lung, and Blood Institute (NHLBI), NIH, Bethesda, MD 20814, USA
*Corresponding Author: Xiangning Bu, Ph.D. National Heart, Lung, and Blood Institute (NHLBI), NIH, Bethesda, MD, 20852, USA. Tel: +1 (301) 594-7359;
Background: The cause of leukemia, the most common type of cancer, remains unknown. Genetic studies have reported more than a thousand of genes as being linked to the disease.
Methods: A total of 1,093 leukemia candidate genes, identified from leukemia-gene relations data extracted from the ResNet 11 Mammalian database and supported by 6,524 references were evaluated. Four network metrics were used to evaluate individual gene potential relevance to leukemia. Gene-set enrichment, sub-network enrichment, and network-connectivity analyses were conducted on gene attributes. An expression dataset of 71 leukemia patients, and 76 healthy controls, was employed for validation.
Results: A total of 952 out of 1,093 genes were enriched in 100 pathways (p < 3.3e-20), demonstrating strong gene-gene interaction. A network metrics analysis revealed 5 genes (TP53, CTNNB1, AKT1, TNF, and RARA), as measured by both functional diversity and replication frequency, as the top leukemia candidates. Validation, using expression data, showed that the 1,093 genes, as a whole, and the top genes, as identified by the proposed metrics, were efficient in distinguishing leukemia patients from controls (maximum classification ratio = 95.3 % with permutation p-value = 0.0054).
Conclusion: The genetic causes of leukemia are linked to a genetic network composed of a large number of genes. This network, together with the network metrics provided in this study, could provide a basis for further molecular studies in the field.
Leukemia is a group of cancers, usually originating in bone marrow, which result in great numbers of abnormal white blood cells. It is the most common type of childhood cancer, even though approximately 90 % of all leukemia cases are in adults [1]. The precise causes of leukemia remain unknown. Inherited and environmental factors are both thought to be involved [2].
More than a thousand genes related to leukemia, many of which suggested as potential biomarkers for the disease, such as FLT3, WT1, TET2, and KRAS, have been reported [3-5]. Several genes, such as IL2 and CSF3, have been studied in clinical trials [6, 7]. Many articles have reported genetic changes, and gene quantitative changes, in leukemia [8, 9]. Both increased, and decreased, gene expression levels/activities have been observed [10-12]. Many genes have been reported to influence leukemia pathogenic development via unknown mechanisms [13].
We found no study reporting a systematic evaluation of the quality, and strength, of these reported genes as a functional network/group in the underlying biological process of leukemia. This study, instead of focusing on specific genes, attempts to provide a comprehensive view of the genetic-map, and use gene set enrichment analysis (GSEA) and sub-network enrichment analysis (SNEA) to study the underlying functional profiles of the genes identified [14]. The hypothesis is that leukemia genes are functionally linked to each other and co-regulate leukemia’s pathogenic development via multiple pathways.
The study workflow was as follows: 1) acquisition of a leukemia-gene relation dataset and identification of leukemia candidate genes; 2) enrichment analysis of the identified genes to study their pathogenic significance to leukemia; 3) network metrics analysis to identify genes having specific significance; 4) network connectivity analysis (NCA) to test functional associations between the reported genes; and, 5) validation using an independent gene expression data set.
2.1 Leukemia-Gene relation Data AcquisitionLeukemia-gene relation data were extracted from the Pathway Studio ResNet® Mammalian database updated as of May 2016. The genes identified were used as the candidate network nodes/genes.
The ResNet® Mammalian database was a part of the Pathway Studio ResNet Databases. This is a group of real-time updated network databases and includes: curated signaling; cellular process and metabolic pathways; ontologies and annotations; and, molecular interactions and functional relationships extracted from the 35M + references covering the entire PubMed abstract and Elsevier full text journals. It is updated weekly. The ResNet® Mammalian database contains information for more than 6,500,000 functional relationships for humans, rats, and mice and is linked to all of the original literature sources. The database includes: 1) 142,270 proteins; 2) 106,732 small molecules; 3) 8,863 cell processes; 4) 15,911 diseases; 5) 5,038 functional classes; 6) 4,387 Clinical parameters; 7) 1,983 pathways; 8) 559 complexes; and, 9) 767 cells. (ResNet databases, http: //pathwaystudio. gousinfo.com/ResNetDatabase.html) .
2.2 Literature metrics analysisThere were 2 scores proposed for each gene-disease relationship as a literature metrics analysis.
The reference number underlying a gene-disease relationship as the gene reference score (RScore) is defined by Eq.(1).
RScore = The number of references underlying a relationship (1)
The earliest publication age of a gene-disease relationship is the gene age score (AScore) and is defined by Eq.(2)
AScore = max1≤i≤n ArticlePubAgei (2)
where n is the total number of references supporting a gene-disease relation, and
ArticlePubAge = Current date-Publication date + 1 (3)
2.3 Enrichment metric analysisGiven a disease associated with a set of genetic pathways ℛ the gene-wise enrichment score (EScore) for the kth gene, within a gene set of size n, is defined in Eq. (4) as
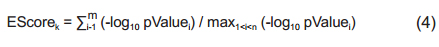
where pValue i is the enrichment score of the ith pathway with the gene set; m ∈ ℛ is the number of pathways including kth the gene. The PScore for the gene, m, is defined as
PScorek = The number of pathways that form ℛ including the kth gene (5)
The PScore presents how many disease-related pathways were associated with the genes. The EScore shows involved pathway significance.
2.4 Enrichment analysisGSEA and SNEA [15] were performed on: 1) entire gene list (1,093 genes); and 2) 2-subgroups with the highest metric scores to better understand any underlying functional profiles and gene pathogenic significance. An NCA was conducted on the two 2-subgroups.
2.5 Validation using gene expression dataThe hypothesis is that significant leukemia candidate gene-gene sets should be a factor in distinguishing leukemia patients from healthy controls. A Euclidean distance-based multivariate classification [16] on an expression dataset, followed by a leave-one-out (LOO) cross validation, using the overall gene set and the sub-sets selected by different scores as tentative markers was performed to evaluate the effectiveness of the selected genes and the proposed metrics.
A permutation of 5,000 runs was then conducted to test the hypothesis that a randomly selected gene set of the same size could lead to equal, or better, classification accuracy.
Expression data from 147 subjects, including samples from 71 chronic, lymphocytic leukemia (CLL) tumors, and 76 sorted CD19pos B cells from healthy donors (NCBI GEO: GSE50006), with 1,031 genes overlapped with the candidate leukemia gene-pool identified within the leukemia-gene dataset.
There were 1,093 leukemia candidate genes identified from the leukemia-gene relation data set. They are supported by 6,524 articles (Supplementary Material 1).There were 994 (90.94 %) which presented a regulation relationship to the disease; 133 (12.17 %) a genetic change; 61 (5.58 %) a quantitative change; 52 (4.76 %) cell expression; 20 (1.83 %) with Biomarker, 17 (1.56 %) with clinical trial, and 5 (0.46 %) with state changes. There are 148 (13.54 %) genes that have been reported to have multiple relationships with the disease. There were 945 (86.46 %) genes that presented a 1-type relationship to the disease, 113 (10.34 %) with a 2, 31 (2.84 %) with a 3, 3 (0.27 %) with a 4, and 1 (0.09 %) with a 5. For a detailed definition and description of these relation types mentioned above, refer to the ‘Relations: Definitions and Annotations’ section at http://pathwaystudio. Gousinfo. com/ ResNet Database. html. Genes with ‘m*’ and ‘r*’ are genes identified in mice and rats, respectively.
 Fig. 1 Gene Relation Type Distribution of the 1,093 Genes
Fig. 1 Gene Relation Type Distribution of the 1,093 Genes
The publication date distribution for these 6,531 articles appears in Fig. 2 (a). Novel genes are reported in each year. These have an average publication age of 6.0 years indicating that most were published recently. Publication date distributions for most of the articles underlying the 1,093 genes were similar (Fig. 2 (b)).
 Fig. 2 Histogram of publications reporting gene-disease relationships between leukemia and the 1,093 Genes.
Fig. 2 Histogram of publications reporting gene-disease relationships between leukemia and the 1,093 Genes.
(a) Number of articles published by year; (b) Gene-wise publication date distribution of the supporting references, with mean marked as red star.
Of these 1,093 genes, 31 were reported in the period January through April within this year 2016 (Table 1). Table 1 also lists the top 31 genes with the highest RScores (in descending order). Full results appear in Supplementary Material 1.
 Table 1. Top 31 Genes with Reported Associations to Leukemia Ranked by Different Scores
Table 1. Top 31 Genes with Reported Associations to Leukemia Ranked by Different Scores
This section presents GSEA and SNEA results for 3 different groups: the 1,093 genes, and both gene groups listed in Table 1.
3.3.1 Enrichment analysis of the 1,093 genesTable 2 presents the top 20 pathways/groups enriched with 857/1,093 genes (p- values < 3.7e-41). A complete list of the 100 pathways/gene sets enriched with 952/1,093 genes (p-value < 3.3e-20) appears in Supplementary Material 2.
Among the 100 pathway/groups enriched, 6 related to cell apoptosis (345/1,093 genes), 9 to cell growth and proliferation (366/1,093 genes), 6 to protein phosphorylation (201/1,093 genes), 3 to the immune system (319/1,093 genes), 11 to transcription factors (449/1,093 genes), 8 to protein kinase (234/1,093 genes), and 2 to neuronal systems (257/1,093 genes).
 Table 2. Molecular Function Pathways/Groups Enriched by 1,093 Genes Reported
Table 2. Molecular Function Pathways/Groups Enriched by 1,093 Genes Reported
Note: The Jaccard similarity (Js) is a statistic used for comparing the similarity and diversity of sample sets, which is defined by Js (A,B) = (A ∩ B) / (A ∪ B) , where A and B are two sample sets.
The top 10 disease-related sub-networks enriched with a p-value < 5E-254 appear in Table 3. Complete results appear in Supplementary Material 3.
 Table 3. Sub-networks Enriched by the 1,093 Genes
Table 3. Sub-networks Enriched by the 1,093 Genes
Many of these reported leukemia-related genes are associated with other cancers that were linked to Leukemia, with a large overlap (Jaccard similarity > 0.18).
The GSEA and SNEA results of the top 31 genes listed in Table 1 were compared. The top 10 pathways/sub-networks for the AScore group and the RScore group are presented (Table 4 and Table 5). Complete results appear in Supplementary Material 2 and 3.
Using a p-value threshold (p < 1E-4), the 31 genes with top AScores were enriched within 10 pathways/groups. The RScore group score was 153.
The top 10 pathways enriched with the 31 genes from the AScore and RScore groups appear in Table 4. A complete listing of these pathways/gene sets appears in Supplementary Material 2.
 Table 4. Pathways/Groups Enriched by 31 Genes with the Highest AScore and RScore
Table 4. Pathways/Groups Enriched by 31 Genes with the Highest AScore and RScore
Genes with the top AScores and those with the top RScores enriched different groups of pathways with different p-values (AScore group: 7.62E-07 - 9.23E-05; RScore group: 1.12E-17 - 2.71E-10). This suggests that the newly-reported genes are both functionally distinct, and are less significant, compared to those most frequently reported.
It was observed that 4/10 pathways/gene sets enriched by the RScore group (Table 4) in Table 2 also appear in the top 20 pathways/groups enriched with 857/1,093 genes. The AScore group had none.
Results from the SNEA analysis consist of an enrichment analysis against disease sub-networks. Table 5 presents the top 10 disease-related sub-networks enriched by the top 31 genes from the AScore group and the RScore group, respectively. Complete results appear in Supplementary Material 3.
 Table 5. SNEA Results by 31 Genes with the Highest AScore and RScore
Table 5. SNEA Results by 31 Genes with the Highest AScore and RScore
From Table 5, both groups enriched other cancer related sub-networks. Enrichment p-values of the RScore group were much more significant than those of the AScore group (NScore group: 4.90E-05 - 2.72E-04; RScore group: 1.41E-52 - 9.32E-41), and have greater Jaccard similarities.
3.4 Connectivity analysisAn NCA was performed on the top 31 genes with the highest RScores and AScores (from Table 1) being used to generate gene-gene interaction networks. Results showed that, for the RScore group, there were 441 connections among the 31 genes, which has significant literature support. In contrast, genes within the AScore group demonstrated only 15 relations among 19/31 genes (Fig. 3 (b)) with 12 genes showing no direct relations with other genes in the group (Fig. 3 (b); highlighted in green). This observation was consistent with the GSEA and SNEA, and suggests that genes with the lowest AScore were not as functionally close to each other as the RScore group.
 Fig. 3 Connectivity Networks built by 31 Genes from Different Groups.
Fig. 3 Connectivity Networks built by 31 Genes from Different Groups.
(a) 31 genes from RScore group; (b) 31 genes from AScore group. The networks were generated using Pathway Studio. Unrelated genes appear in green.
Using GSEA, two biological metrics, EScore and PScore were generated for each gene. The PScore value represents how many leukemia associated pathways involved the gene. The EScore shows pathway significance.
A correlation analysis using averaged metric values of all 1,093 genes at a group level was conducted to compare the EScore and PScore with the two literature metrics (Fig. 4 (a)). A group size of 36 genes was used. The 1,093 genes were sorted by RScore, then averaged by each type of metrics values using a moving window of length 36.
Results showed that the average scores strongly correlate, especially for the top ones. (Fig. 4 (a) and Table 6). Group-wise PScore and EScore were extremely correlated (p=0.99) .
 Fig. 4 Comparison of Different Metrics Ranking the 1,093 Genes.
Fig. 4 Comparison of Different Metrics Ranking the 1,093 Genes.
(a)Comparison of the average metrics values with gene set size of 36; (b) A Venn diagram of the top 31 genes selected by different metrics.
 Table 6. Pearson Correlation Coefficients between Different Metrics
Table 6. Pearson Correlation Coefficients between Different Metrics
In addition to the group-wise correlations analysis, a cross-analysis of the top 31 genes selected using different scores was performed and is presented in a Venn Diagram. (Fig.4 (b)) (Oliveros, 2007-2015).
There was a significant overlap between the PScore group and the EScore group (28/31). These 28 genes related to most pathways that were significantly enriched. Additionally, 5 genes were identified as being in overlapping EScore, PScore, and RScore groups, including TP53, CTNNB1, AKT1, TNF, and RARA (RScore: 58.80 ± 11.17 references, PScore: 34.00 ± 2.55 pathways) (Fig. 4 (b)). There were 23 genes observed in both the PScore group and the EScore group, but not in the RScore group. This included: RELA, JUN, EGFR, SRC, J AK2, HIF1A, TGFB1, HDAC1, STAT3, IL1B, PTK2, FYN, LCK, LYN, MAPK3, IL6, MAPK1, EP300, CREB1, ERBB2, PDGFRB, GSK3B, and KDR. These genes played roles within multiple significant pathways with leukemia (32.04.28 pathways). Although they were older (AScore: 12.486.71 years) and were not frequently replicated (RScore: 10.04 ± 8.94 references), the results suggest that they are worthy of further study.
3.6 Validation using expression dataSignificant leukemia candidate gene-gene sets were hypothesized as contributing to being able to distinguish leukemia patients from healthy controls. If the selected gene set (1,093 genes) and the top genes selected by the proposed metric scores are significant to leukemia pathogenesis, then they should lead to significant higher classification accuracies when compared to randomly selected gene sets. To test this hypothesis that the 1,093-gene-pool and the 4 proposed metrics are effective, classification and leave-one-out (LOO) cross validation was conducted on a gene expression dataset (NCBI GEO: GSE50006). This was followed by a 5,000-run permutation test.
The 1,093 genes were ranked by different metric scores. The top ( = 1, 2, …) genes were then used as input variables for classification and LOO cross validation. LOO results using different number of genes, with the maximum classification ratios (maxCRs) marked at the position of corresponding number of genes appear in Fig. 5 (See Table 7).
 Fig. 5 Comparison of LOO Cross Validations Metrics.
Fig. 5 Comparison of LOO Cross Validations Metrics.
The top genes selected by different scores can lead to the highest classification accuracies, adding more variable/genes with lower score may not necessarily help, which demonstrates the effectiveness of the proposed metrics (Fig. 5). All four groups (RScore, AScore, PScore, and EScore), obtained the highest scores of 94.6 %, 95.3 %, 92.1 % and 92.1 %, respectively, with a relatively small number of genes. All the permutation p-values of these groups passed the 0.05 threshold. The top 33 genes, by AScore, led to the highest CR (95.3 %), with a permutation p-value of 0.0054. Employing all matched 1,031/1,093 genes, resulted in 92.1 % CR which was reached with a permutation p-value of 0.037. This suggests that the majority of the 1,093 genes were effective for leukemia prediction. The results of LOO cross-validation and permutation approaches for different gene sets appear in Table 7.
 Table 7. Permutation Test for Top Genes Corresponding to the Highest CRs
Table 7. Permutation Test for Top Genes Corresponding to the Highest CRs
This study proposed 4 network metrics to evaluate the 1,093 candidate genes within a genetic network for leukemia. It employed an independent gene expression data set to validate their efficiencies. GSEA, SNEA, and NCA were also used to study the pathogenic significance of these candidate genes in the disease.
The 1,093 genes identified were not equal in terms of publication frequency (RScore), novelties (AScore) , or the functional diversity (EScore). Using the proposed quality metrics scores, the genes may be ranked according to different needs/significance and the top ones selected for further analysis (see Supplementary Material 1). Some frequently replicated genes (with a high RScore) also demonstrate high EScore and PScore, such as TP53, CTNNB1, AKT1, TNF, and RARA (see Fig. 4 (b)). These genes have an average support of 58.80 ± 11.17 references, and were connected to multiple, significantly-enriched, pathways (34.00 ± 2.55). The results suggest that these genes are biologically significant in the disease.
There were 23 genes observed in both the PScore group and the EScore group (Fig. 4 (b)) which were not in the RScore group. Although they were older (AScore: 12.48 ± 6.71 years) and were not frequently replicated (10.04 ± 8.94 references), the results suggest that they merit further study. One example, the gene RELA, regulation of NF-kappa B transcription factor activity (0051092); aging (GO: 0016280); liver development (GO: 0001889); negative regulation of apoptotic process (GO: 0006916); positive regulation of cell proliferation (GO: 0008284); transcription factor complex (GO: 0005667); innate immune response (GO: 0002226); and, protein kinase binding (GO: 0019901) [17-26]. This suggests that these genes may play significant roles in leukemia pathology and, thus, merit additional study.
The results demonstrate that most genes identified in this study were included in previously-implicated leukemia pathways. This included 6 cell apoptosis pathways, 9 cell growth and proliferation pathways, 11 transcription factor pathways, 7 protein phosphorylation related pathways, 3 immune system pathways, 8 protein kinase related pathways, and 2 neuronal system pathways [21-27]. We hypothesize that the majority of these literature-reported genes, especially those identified from significantly enriched pathways, should be functionally linked to leukemia. Although there may be false positives from the separate studies in the publications, it is less likely that a numerous group of genes were falsely perturbed [14].
When members of a gene set exhibit strong cross-correlation, GSEA boosts the signal-to-noise ratio making it possible to detect modest changes in individual genes [14]. The NCA analysis showed that many of the frequently reported genes related to leukemia are functionally associated with one another (Fig. 3). This is supported by hundreds of scientific reports. It should be noted that 952/1,093 were included in the top 100 pathways enriched (p-value < 3.3e-020), and that 857/1,093 in the top 20 pathways appear in Table 2 (p-value < 3.7e-041) .
If “functionally related” is defined as co-existence within the same genetic pathway, then 87.1 % of the 1,093 genes are functionally related. The results indicate that these functionally-linked genes are more likely to be true discoveries than noise (false positives). It is less likely that these functionally-related genes were falsely identified than a single gene.
A Sub-Network Enrichment Analysis (SNEA) was performed which provided high confidence levels when interpreting experimentally-derived genetic data against a background of previously-published results (Pathway Studio Web Help). SNEA results revealed that many of the 1,093 genes ( > 90 %) have also been identified as causal genes for other health disorders such as, breast cancer, hepatocellular carcinoma, and lung cancer, all of which have strong associations with leukemia [28-30].
A LOO cross-validation and permutation process using a gene expression data set (NCBI GEO: GSE50006) identified several significant gene combinations by using different scores, which generated the highest CRs. Permutation results showed that the top genes as determined by these four scores, as well as the 1,031/1,093 genes, were effective in predicting leukemia (p-value < 0.05). This indicates the effectiveness of the proposed metric scores. The top 33 genes selected by AScore reached the highest CR, 95.3 %, with a permutation p-value of 0.0054. This suggests that the genes identified in the earliest stage of leukemia genetic studies play a significant role in leukemia prediction.
This study has several limitations that should be considered in future work. The 1,093 genes were identified from leukemia-gene relation data extracted from the Pathway Studio ResNet database. Although supported by 6,524 articles, it is possible that some leukemia-gene relationships may have not been identified. The 4 proposed metrics were effective in selecting the top genes for leukemia prediction. Further network analysis with more experimental data may extract additional useful features for identifying biologically significant genes.
Leukemia is a complex, genetically-caused, disease with the genetic causes linked to a large gene network. Integrating network gene-disease relation data and experimental data, with GSEA, SNEA, and NCA, may provide and effective approach to identifying potential target genes. This study provides an overview map for the current field of genetic research of leukemia, which could be used as the basis in future biological/genetic studies.
The authors declare no conflicts of interest.
This research received no specific grants from funding agencies in the public, commercial, or not-for-profit sectors.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
Supplementary File 1: Zip File
Xiang X, Foster P, Zhang X, Bu X. Leukemia Candidate Genetic Markers-An Evaluation. Med One. 2016; 1(4): 6; https://doi.org/10.20900/mo.20160019

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions



